Pedigree Analysis and Genetic Diversity in R
Source:vignettes/pedigree-analysis.Rmd
pedigree-analysis.RmdThis vignette presents a practical workflow for pedigree analysis and
genetic diversity assessment in R with visPedigree. It
emphasizes the interpretation of the main indicators used in breeding
and conservation genetics.
The discussion is organized around five questions:
- How complete and deep is the pedigree?
- How long is the generation cycle?
- Has genetic diversity been eroded by unequal founder use, bottlenecks, or drift?
- How large is the effective population size under different definitions?
- Are relationship, inbreeding, subpopulation structure, and gene flow under control?
1. Setup and Data Preparation
Different package datasets are used for different analytical tasks.
-
deep_pedis useful for pedigree depth and diversity summaries. -
big_family_size_pedcontains aYearcolumn and is suitable for generation intervals. -
small_pedis convenient for relationship examples. -
inbred_pedis convenient for inbreeding and classification examples.
library(visPedigree)
library(data.table)
#>
#> Attaching package: 'data.table'
#> The following object is masked from 'package:base':
#>
#> %notin%
data(deep_ped, package = "visPedigree")
data(big_family_size_ped, package = "visPedigree")
data(small_ped, package = "visPedigree")
data(inbred_ped, package = "visPedigree")
tp_deep <- tidyped(deep_ped)
tp_small <- tidyped(small_ped)
tp_inbred <- tidyped(inbred_ped)2. Pedigree Overview with pedstats()
pedstats() provides a compact structural summary with
three components:
-
$summary: pedigree size and parental structure. -
$ecg: pedigree completeness and ancestral depth. -
$gen_intervals: generation intervals, only when a usable time column is available.
Here deep_ped has no explicit birth-date column.
Accordingly, pedstats() returns $summary and
$ecg, whereas $gen_intervals remains
NULL.
stats_deep <- pedstats(tp_deep)
stats_deep$summary
#> N NSire NDam NFounder MaxGen
#> <int> <int> <int> <int> <int>
#> 1: 4399 483 554 138 13
tail(stats_deep$ecg)
#> Ind ECG FullGen MaxGen
#> <char> <num> <num> <num>
#> 1: K110997Q 7.616211 5 12
#> 2: K110997Z 6.722656 5 12
#> 3: K110998Q 4.606445 2 12
#> 4: K110998Z 6.417969 5 12
#> 5: K110999Q 7.345215 5 12
#> 6: K110999Z 6.875977 5 12
stats_deep$gen_intervals
#> NULL
# Visualize ancestral depth (Equivalent Complete Generations)
plot(stats_deep, type = "ecg", metric = "ECG")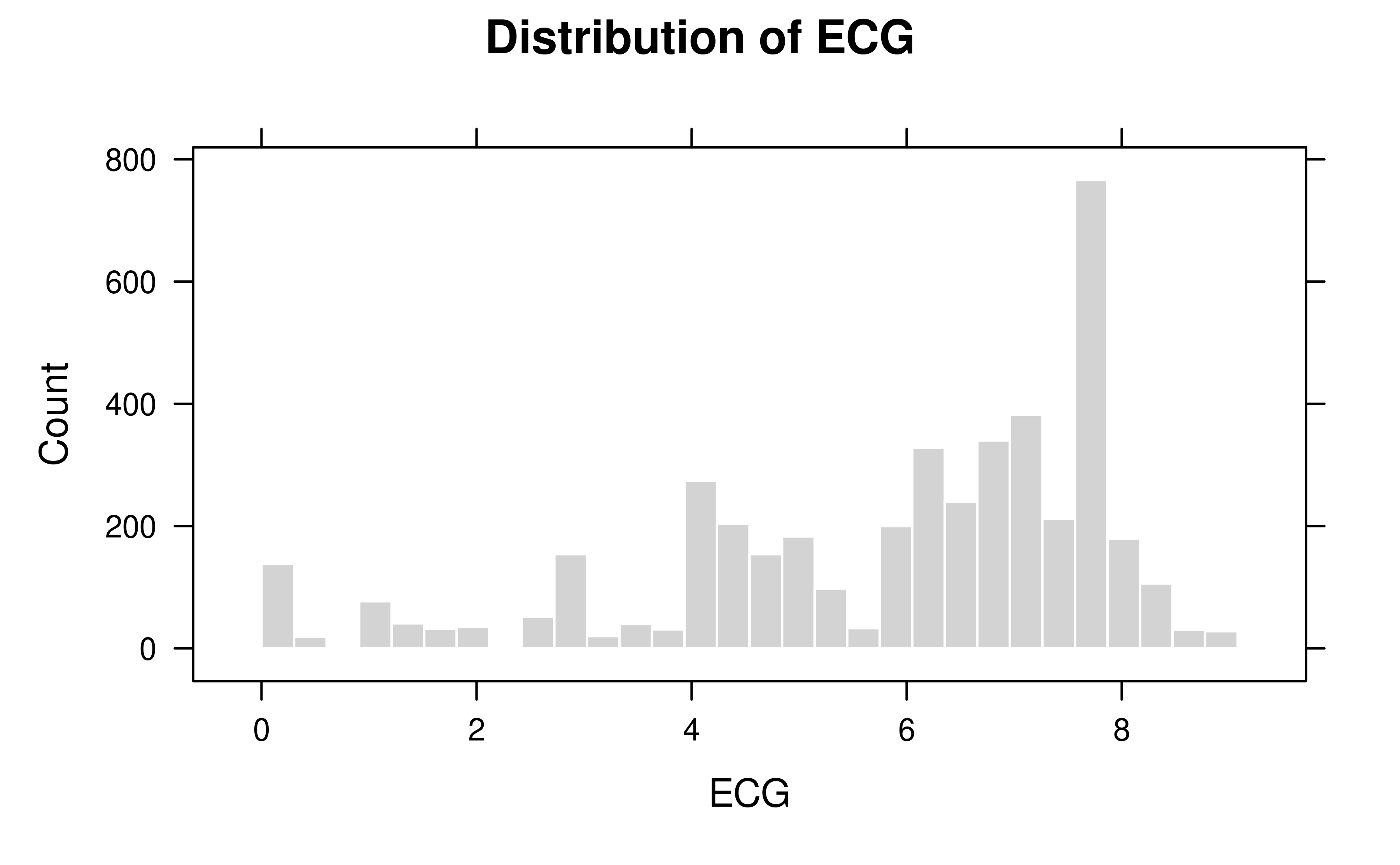
Pedigree depth and pedigree time are related but distinct quantities.
The former is addressed by pedecg(), whereas the latter is
addressed by pedgenint().
3. Pedigree Completeness with pedecg()
Equivalent complete generations (ECG) summarize the amount of ancestral information available for each individual:
where is the number of generations between individual and ancestor . ECG increases with both pedigree depth and pedigree completeness.
In practice:
-
ECGcombines depth and completeness. -
FullGencounts how many fully known ancestral generations exist. -
MaxGenrecords the deepest known ancestral path.
ECG is especially useful because it provides the depth
adjustment required by realized effective population size estimators
based on inbreeding or coancestry.
ecg_deep <- pedecg(tp_deep)
ecg_deep[order(-ECG)][1:10]
#> Ind ECG FullGen MaxGen
#> <char> <num> <num> <num>
#> 1: K110034Q 9.078125 6 12
#> 2: K110052L 9.078125 6 12
#> 3: K110060H 9.078125 6 12
#> 4: K110069Z 9.078125 6 12
#> 5: K110097Q 9.078125 6 12
#> 6: K110118M 9.078125 6 12
#> 7: K110131M 9.078125 6 12
#> 8: K110138M 9.078125 6 12
#> 9: K110155M 9.078125 6 12
#> 10: K110165M 9.078125 6 124. Generation Intervals with pedgenint()
Generation interval is the age of a parent at the birth of its offspring:
pedgenint() estimates this quantity for seven pathway
summaries:
-
SS,SD,DS,DD: sex-specific gametic pathways. -
SO,DO: sex-independent sire-offspring and dam-offspring pathways. -
Average: all parent-offspring pairs combined.
The function accepts Date/POSIXct columns,
date strings, or numeric years. When only an integer year is available,
it is converted internally to YYYY-07-01 as a mid-year
approximation.
tp_time <- tidyped(big_family_size_ped)
genint_year <- pedgenint(tp_time, timevar = "Year", unit = "year")
#> Numeric time column detected. Converting to Date (YYYY-07-01). For finer precision, convert to Date beforehand.
genint_year
#> Pathway N Mean SD
#> <char> <int> <num> <num>
#> 1: Average 280512 1.001093 0.03770707
#> 2: DD 607 1.164398 0.37080261
#> 3: DO 140256 1.001093 0.03770714
#> 4: DS 507 1.196959 0.39776787
#> 5: SD 607 1.164398 0.37080261
#> 6: SO 140256 1.001093 0.03770714
#> 7: SS 507 1.196959 0.39776787
# Visualize generation intervals
# Note: we can also use stats <- pedstats(tp_time, timevar = "Year") followed by plot(stats)
plot(genint_year)
The optional cycle parameter adds GenEquiv,
which compares the observed mean interval with a target breeding
cycle:
Values larger than 1 indicate that the realized generation interval exceeds the target cycle.
genint_cycle <- pedgenint(tp_time, timevar = "Year", unit = "year", cycle = 1.2)
#> Numeric time column detected. Converting to Date (YYYY-07-01). For finer precision, convert to Date beforehand.
genint_cycle[Pathway %in% c("SS", "SD", "DS", "DD", "Average")]
#> Pathway N Mean SD GenEquiv
#> <char> <int> <num> <num> <num>
#> 1: Average 280512 1.001093 0.03770707 0.8342445
#> 2: DD 607 1.164398 0.37080261 0.9703321
#> 3: DS 507 1.196959 0.39776787 0.9974660
#> 4: SD 607 1.164398 0.37080261 0.9703321
#> 5: SS 507 1.196959 0.39776787 0.99746605. Subpopulation Structure with pedsubpop()
Before interpreting diversity or relationship metrics, it is useful to check whether the pedigree forms a single connected population or a mixture of separate components.
pedsubpop() has two modes:
-
by = NULL: summarize disconnected pedigree components. -
by = "...": summarize the pedigree by an existing grouping variable.
ped_demo <- data.table(
Ind = c("A", "B", "C", "D", "E", "F", "G", "H"),
Sire = c(NA, NA, "A", NA, NA, "E", NA, NA),
Dam = c(NA, NA, "B", NA, NA, NA, NA, NA),
Sex = c("male", "female", "male", "female", "male", "female", "male", "female"),
Batch = c("L1", "L1", "L1", "L1", "L2", "L2", "L3", "L3")
)
tp_demo <- tidyped(ped_demo)
pedsubpop(tp_demo)
#> Group N N_Sire N_Dam N_Founder
#> <char> <int> <num> <num> <int>
#> 1: GP1 3 1 1 2
#> 2: GP2 2 1 0 1
#> 3: Isolated 3 0 0 3
pedsubpop(tp_demo, by = "Batch")
#> Group N N_Sire N_Dam N_Founder
#> <char> <int> <int> <int> <int>
#> 1: L1 4 1 1 3
#> 2: L3 2 0 0 2
#> 3: L2 2 1 0 1This is a compact summary tool. When the actual split pedigree
objects are needed for downstream analysis, use
splitped().
6. Diversity Indicators with pediv()
pediv() is the integrated diversity summary. It
combines:
- founder and ancestor contributions (
fe,fa), - founder genome equivalents (
fg), - retained genetic diversity (
GeneDiv), - three effective population size estimates (
Ne).
The gene-origin summaries follow the classical founder/ancestor framework of Lacy (1989) and Boichard et al. (1997), while coancestry-based diversity and effective population size calculations follow Caballero & Toro (2000) and related pedigree-based estimators.
All of these quantities depend on the definition of the
reference population. In the present example, the
reference population is defined as the most recent two generations in
deep_ped.
div_res <- pediv(tp_deep, reference = ref_pop, top = 10, seed = 42L)
#> Calculating founder and ancestor contributions...
#> Calculating founder contributions...
#> Calculating ancestor contributions (Boichard's iterative algorithm)...
#> Calculating Ne (coancestry) and fg...
#> Calculating Ne (inbreeding)...
#> Calculating Ne (demographic)...
div_res$summary
#> NRef NFounder feH fe NAncestor faH fa fafe fg
#> <int> <int> <num> <num> <int> <num> <num> <num> <num>
#> 1: 3471 157 92.0943 64.73344 94 60.1444 44.12033 0.681569 19.17965
#> MeanCoan GeneDiv NSampledCoan NeCoancestry NeInbreeding NeDemographic
#> <num> <num> <int> <num> <num> <num>
#> 1: 0.0260693 0.9739307 1000 124.5124 98.00425 374.2021
div_res$ancestors
#> Ind Contrib CumContrib Rank
#> <char> <num> <num> <int>
#> 1: K500I804 0.05248848 0.05248848 1
#> 2: K60GXQ91 0.04525893 0.09774741 2
#> 3: K60NXQ91 0.04525893 0.14300634 3
#> 4: K600532I 0.04445765 0.18746399 4
#> 5: K700S069 0.03927182 0.22673581 5
#> 6: K900D251 0.03622875 0.26296456 6
#> 7: K700S011 0.02906223 0.29202679 7
#> 8: K700U416 0.02890017 0.32092697 8
#> 9: K700U650 0.02890017 0.34982714 9
#> 10: K500I869 0.02831497 0.37814211 106.1 fe, fa, and fg: what do
they measure?
These three indicators describe diversity loss from complementary angles.
Effective number of founders (fe)
where
is the expected contribution of founder
to the reference population. fe answers the question: if
all founders had contributed equally, how many founders would generate
the same diversity?
Use fe when the main concern is unequal founder
representation.
Effective number of ancestors (fa)
where
is the marginal contribution of ancestor
after removing the contributions already explained by more influential
ancestors. fa is usually smaller than fe when
bottlenecks occurred.
Use fa when the main concern is genetic
bottlenecks caused by a limited set of influential
ancestors.
Founder genome equivalents (fg)
In its simplest interpretation,
where
is the mean coancestry of the reference population. In
visPedigree, fg is estimated from a
diagonal-corrected mean coancestry:
where is the size of the full reference population, is the mean off-diagonal additive relationship among sampled individuals, and is their mean inbreeding coefficient.
Use fg when the main concern is the amount of
founder genome still surviving after unequal use, bottlenecks, and
drift. In practice, fg is often the most
conservative diversity indicator.
Retained genetic diversity (GeneDiv)
GeneDiv is the pedigree-based retained genetic diversity
of the reference population:
where is the same diagonal-corrected mean coancestry used to compute . Because coancestry increases as individuals become more related by descent, decreases towards 0 as the population becomes more uniform. An unrelated, non-inbred population has ; a population of full sibs from two unrelated founders has .
GeneDiv and fg are both derived from
,
but they answer different questions. fg is measured in
“equivalent number of founders” and is most useful for between-programme
comparisons. GeneDiv is a dimensionless proportion and is
easiest to communicate to managers and stakeholders who need a single
intuitive index of diversity retention.
# GeneDiv is in the summary alongside MeanCoan
div_res$summary[, .(fg, MeanCoan, GeneDiv)]
#> fg MeanCoan GeneDiv
#> <num> <num> <num>
#> 1: 19.17965 0.0260693 0.97393076.2 Shannon-entropy effective numbers: feH and
faH
The classical fe and fa are based on
(Hill number order
;
Hill, 1973), which is disproportionately influenced by a few large
contributions. visPedigree also reports the
information-theoretic counterpart at order
,
derived from Shannon entropy (Jost, 2006):
All four metrics satisfy a strict monotone ordering:
where is the total number of founders and is the number of ancestors considered.
Metric guide
| Metric | Order | Sensitivity | What it measures |
|---|---|---|---|
fe |
Dominated by common founders | Effective number of founders if contributions were equalized. Dominated by the largest contributions; many rare founders with small have negligible influence on the metric. | |
feH |
Balanced; sensitive to rare founders | Shannon-entropy effective founders. Counts rare founders more
equitably than
;
always
fe. |
|
fa |
Dominated by common ancestors | Effective number of ancestors accounting for bottleneck structure.
Typically lower than fe. |
|
faH |
Balanced; sensitive to rare ancestors | Shannon-entropy effective ancestors. Less dominated by the single most influential ancestor. | |
fg |
— | Realized coancestry | Founder genome equivalents; the most conservative indicator because it captures drift as well. |
GeneDiv |
— | Retained diversity | Pedigree-based retained genetic diversity: . Values on the scale; higher values indicate more diversity retained relative to the base population. |
Interpreting the ratio
The ratio is a long-tail signal:
- : founder contributions are already concentrated in a small number of individuals; there is no meaningful “hidden” diversity from minor founders.
- : many minor founders still carry genetic material into the reference population but are effectively invisible to the classical metric because their contributions are small. This situation is common early in a breeding programme when the pedigree is still shallow.
The analogous ratio diagnoses the same effect at the ancestor level: a large ratio means that the bottleneck signal from the dominant ancestor is masking genuine contributions from many secondary ancestors.
Management implications
Use the two ratios together to set priorities:
- If is large but itself is already high, the programme has good founder coverage; no immediate action needed.
- If is large and is low, minor founders carry material that could be mobilized — consider deliberately increasing their representation in the next generation.
- If is large, secondary ancestors are still present but marginalized; diversifying sire selection could rebalance the gene pool before their lineages are lost entirely.
# Shannon metrics are included in pediv() output
div_res$summary[, .(NFounder, feH, fe, NAncestor, faH, fa)]
#> NFounder feH fe NAncestor faH fa
#> <int> <num> <num> <int> <num> <num>
#> 1: 157 92.0943 64.73344 94 60.1444 44.12033
# The ratio feH/fe reveals long-tail founder diversity
div_res$summary[, .(rho_founder = feH / fe, rho_ancestor = faH / fa)]
#> rho_founder rho_ancestor
#> <num> <num>
#> 1: 1.42267 1.36319
# pedcontrib() provides the same metrics via its summary
contrib_res <- pedcontrib(tp_deep, reference = ref_pop, mode = "both")
#> Calculating founder contributions...
#> Calculating ancestor contributions (Boichard's iterative algorithm)...
contrib_res$summary[c("n_founder", "f_e_H", "f_e", "n_ancestor", "f_a_H", "f_a")]
#> $n_founder
#> [1] 157
#>
#> $f_e_H
#> [1] 92.0943
#>
#> $f_e
#> [1] 64.73344
#>
#> $n_ancestor
#> [1] 94
#>
#> $f_a_H
#> [1] 60.1444
#>
#> $f_a
#> [1] 44.120337. Diversity Dynamics over Time with pedhalflife()
pediv() provides a static snapshot of diversity. When
multiple time points (generations, years, etc.) are available,
pedhalflife() tracks how
,
,
and
evolve and fits a log-linear decay model to quantify the rate of
diversity loss.
The total loss rate is decomposed into three additive components that each have a distinct biological meaning:
| Component | Symbol | Source of loss |
|---|---|---|
| Foundation | Unequal founder contributions | |
| Bottleneck | Overuse of key ancestors | |
| Drift | Random sampling loss |
The diversity half-life is the number of time units for to halve:
hl <- pedhalflife(tp_deep, timevar = "Gen")
#> Calculating diversity across 13 time points...
print(hl)
#> Information-Theoretic Diversity Half-Life
#> -----------------------------------------
#> Total Loss Rate (lambda_total): 0.128778
#> Foundation (lambda_e) : 0.034626
#> Bottleneck (lambda_b) : 0.025217
#> Drift (lambda_d) : 0.068935
#> -----------------------------------------
#> Diversity Half-life (T_1/2) : 5.38 (Gen)
#>
#> Timeseries: 13 time pointsThe printed half-life shows the unit in parentheses —
(Gen) here — taken directly from the timevar
argument. The full object exposes three components:
-
hl$timeseries— per-time-point table (see column guide below). -
hl$decay— single-row table withLambdaE,LambdaB,LambdaD,LambdaTotal, andTHalf. -
hl$timevar— thetimevarstring passed to the call.
7.1 Column guide for $timeseries
The cascade rests on a simple algebraic identity:
Because OLS is linear, the slope of versus time equals the sum of the slopes of the three right-hand terms. This guarantees exact additivity: .
| Column | Formula | What it measures |
|---|---|---|
lnfe |
Log effective-founder diversity. Declines when founder contributions become more unequal over time. | |
lnfa |
Log effective-ancestor diversity. Lies below lnfe
whenever bottleneck ancestors have been over-used. |
|
lnfg |
Log founder-genome equivalents. The most comprehensive diversity signal; integrates founder use, bottlenecks, and drift. | |
lnfafe |
Bottleneck gap. When this ratio decreases over time, a small number of ancestors dominate the gene pool even beyond what unequal founder use would predict. | |
lnfgfa |
Drift gap. Captures random allele loss due to finite population size. Because always, this term is and becomes more negative as genetic drift accumulates. | |
TimeStep |
numeric | OLS time axis (same as Time for numeric
timevar; sequential indices otherwise). |
7.2 Interpreting the components
The negative OLS slope of each log column gives the corresponding loss rate. All three terms have a distinct policy implication:
(Foundation) — rate of increase in founder-contribution inequality. A large suggests that founder representation is still diverging, possibly because some founders are reaching later generations through many more breeding pathways than others. Remedy: broaden founder use in the breeding programme.
(Bottleneck) — rate at which a few key ancestors capture an ever-larger share of the gene pool beyond what founder inequality alone explains. A large points to systematic over-selection of certain families or sires. Remedy: restrict the number of offspring per high-ranking animal; rotate breeding males.
(Drift) — rate of random allele loss in finite populations. This component is always present and increases with small effective population size. It is the only component that cannot be reversed by changing selection decisions; it can only be slowed by maintaining a larger breeding population or by cryopreservation of genetic material.
When (as in the example above), drift is the dominant driver of diversity loss and should be the primary management target.
7.3 Interpreting
is the number of time units (generations, years, etc.) required for to fall to half its current value, assuming the observed decay rate continues. It is a single headline figure that combines all three loss channels.
As a rough benchmark: a half-life shorter than five to ten generations is generally considered a cause for concern in conservation and aquaculture genetics. Breed registries and conservation programmes often use as a threshold indicator for triggering corrective management.
hl$timeseries
#> Time NRef fe fa fg lnfe lnfa lnfg
#> <int> <int> <num> <num> <num> <num> <num> <num>
#> 1: 1 138 138.00000 138.00000 138.000000 4.927254 4.927254 4.927254
#> 2: 2 96 81.55752 74.02410 57.242236 4.401309 4.304391 4.047292
#> 3: 3 74 69.53651 55.45316 36.204959 4.241852 4.015539 3.589196
#> 4: 4 91 33.07439 22.47218 18.505028 3.498759 3.112278 2.918042
#> 5: 5 95 58.21114 26.60280 17.148880 4.064077 3.281016 2.841933
#> 6: 6 119 20.87729 13.46723 9.709381 3.038662 2.600259 2.273093
#> 7: 7 37 40.12238 24.89445 10.016763 3.691934 3.214645 2.304260
#> 8: 8 41 43.07344 23.20121 9.927020 3.762907 3.144204 2.295260
#> 9: 9 51 43.45747 25.55284 11.468733 3.771783 3.240749 2.439624
#> 10: 10 78 58.80040 35.70455 14.967166 4.074149 3.575278 2.705859
#> 11: 11 108 49.32442 33.37264 14.920611 3.898419 3.507737 2.702744
#> 12: 12 209 51.27256 33.93121 14.728712 3.937156 3.524335 2.689799
#> 13: 13 3262 65.47682 44.33880 19.682156 4.181696 3.791860 2.979712
#> lnfafe lnfgfa TimeStep
#> <num> <num> <num>
#> 1: 0.0000000 0.0000000 1
#> 2: -0.0969179 -0.2570986 2
#> 3: -0.2263131 -0.4263427 3
#> 4: -0.3864809 -0.1942358 4
#> 5: -0.7830602 -0.4390836 5
#> 6: -0.4384028 -0.3271666 6
#> 7: -0.4772897 -0.9103847 7
#> 8: -0.6187023 -0.8489440 8
#> 9: -0.5310341 -0.8011241 9
#> 10: -0.4988704 -0.8694193 10
#> 11: -0.3906828 -0.8049930 11
#> 12: -0.4128205 -0.8345365 12
#> 13: -0.3898361 -0.8121477 13To focus on a specific time window, pass the desired values to the
at argument:
hl_recent <- pedhalflife(tp_deep, timevar = "Gen",
at = tail(sort(unique(tp_deep$Gen)), 4))
#> Calculating diversity across 4 time points...
print(hl_recent)
#> Information-Theoretic Diversity Half-Life
#> -----------------------------------------
#> Total Loss Rate (lambda_total): -0.077122
#> Foundation (lambda_e) : -0.036138
#> Bottleneck (lambda_b) : -0.030497
#> Drift (lambda_d) : -0.010487
#> -----------------------------------------
#> Diversity Half-life (T_1/2) : NA
#>
#> Timeseries: 4 time pointsPlotting the object shows the log-linear decay of each diversity metric:
plot(hl, type = "log")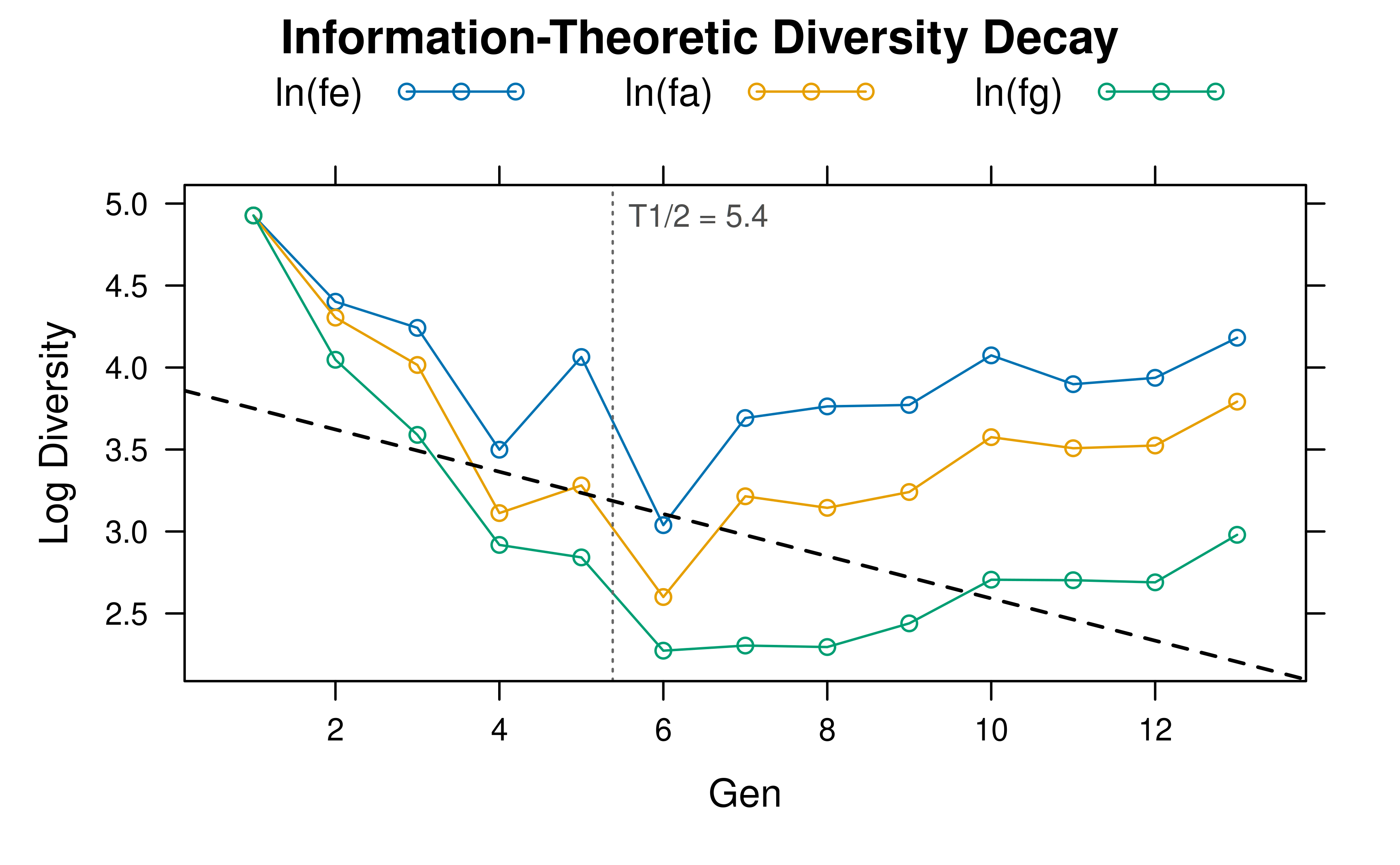
A type = "raw" plot shows the equivalent numbers on the
original scale:
plot(hl, type = "raw")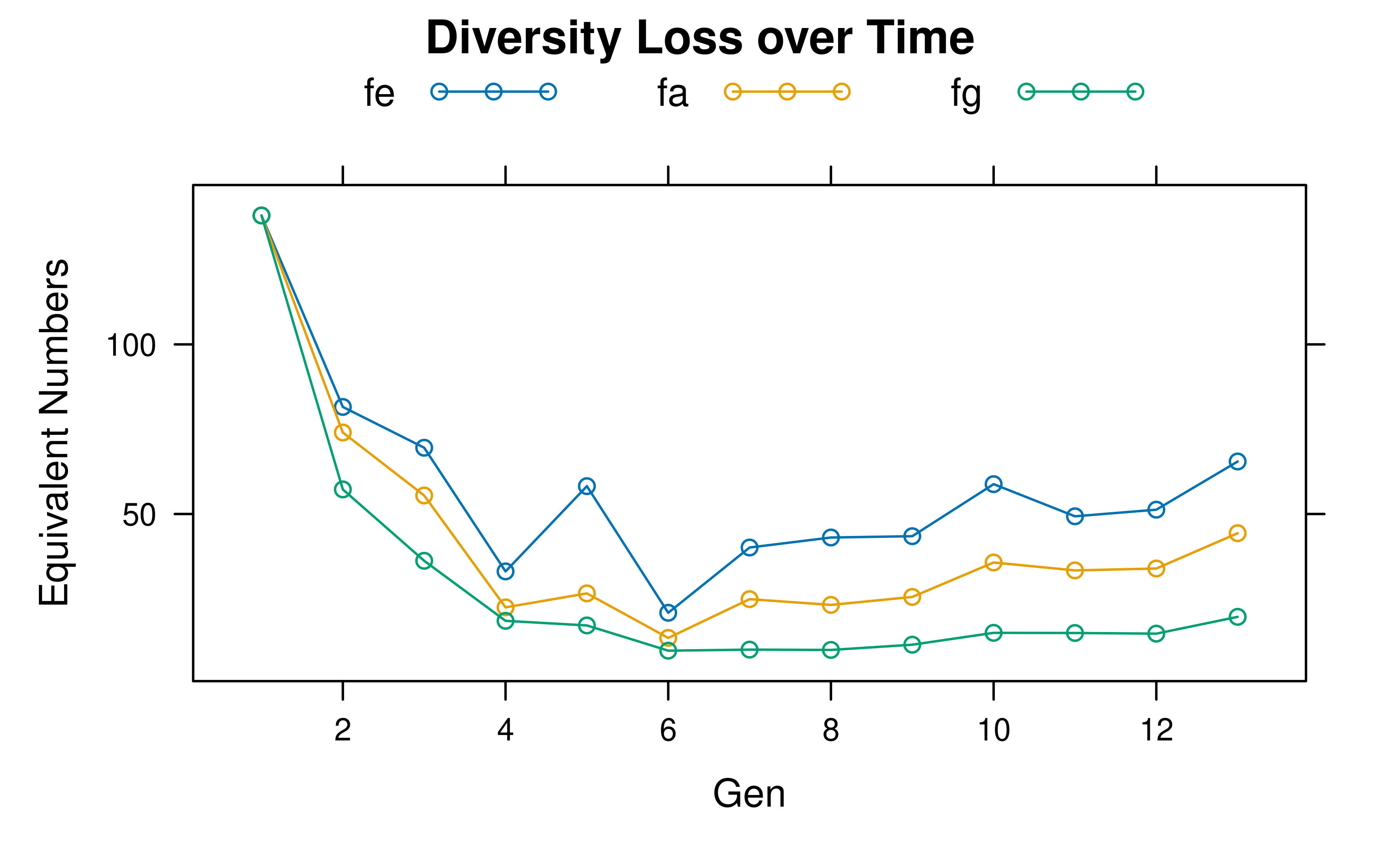
8. Effective Population Size with pedne() and
pediv()
pediv() reports three complementary effective population
size definitions. The pedigree-based estimators follow the
individual-rate approach of Gutiérrez et al. (2008, 2009) and the
coancestry-rate approach of Cervantes et al. (2011). Each addresses a
distinct biological question.
8.1 Demographic Ne
where and are the numbers of contributing males and females. This is the easiest estimate to understand, but it ignores realized pedigree structure, inbreeding, and drift. It is therefore often optimistic.
8.2 Inbreeding Ne
This estimator uses the realized rate of inbreeding (Gutiérrez et
al., 2008, 2009). ECG standardizes individuals with
different pedigree depths. Use this estimate when the primary concern is
the rate of inbreeding accumulation.
8.3 Coancestry Ne
This estimator is based on the rate of coancestry among members of the reference population (Cervantes et al., 2011). Because it captures the accumulation of relatedness before it is fully expressed as realized inbreeding, it is often the strictest and most sensitive warning signal in managed breeding populations.
9. Average Relationship Trends with pedrel()
pedrel() summarizes pairwise relatedness within groups.
Two complementary scales are available via the scale
argument.
9.1 Mean additive relationship
(scale = "relationship")
The default scale returns the mean off-diagonal additive relationship:
where
is the additive relationship and
is the coancestry (kinship coefficient) between individuals
and
.
A higher MeanRel means that individuals within the group
are, on average, more related by descent.
Internally, pedrel() creates an indicator vector
for each group and computes
directly from the pedigree. The off-diagonal sum is obtained from
after subtracting
.
The dense relationship matrix is never materialized, so
compact is now retained only as a backward-compatible no-op
argument.
tp_small$BirthYear <- 2010 + tp_small$Gen
rel_by_gen <- pedrel(tp_small, by = "Gen")
rel_by_gen
#> Gen NTotal NUsed MeanRel Status Message
#> <int> <int> <int> <num> <char> <char>
#> 1: 1 9 9 0.0000000 ok
#> 2: 2 5 5 0.2000000 ok
#> 3: 3 7 7 0.1547619 ok
#> 4: 4 3 3 0.1145833 ok
#> 5: 5 2 2 0.0468750 ok
#> 6: 6 2 2 0.5507812 okThe reference argument lets you focus on a subset of
interest inside each group, such as candidate breeders.
ref_ids <- c("Z1", "Z2", "X", "Y")
pedrel(tp_small, by = "Gen", reference = ref_ids)
#> Warning: pedrel(): 4 of 6 groups returned a non-ok status. Inspect the 'Status'
#> and 'Message' columns. First issue: Gen = 1; Group has less than 2 individuals
#> after applying 'reference'.
#> Gen NTotal NUsed MeanRel Status
#> <int> <int> <int> <num> <char>
#> 1: 1 9 0 NA skipped
#> 2: 2 5 0 NA skipped
#> 3: 3 7 0 NA skipped
#> 4: 4 3 0 NA skipped
#> 5: 5 2 2 0.0468750 ok
#> 6: 6 2 2 0.5507812 ok
#> Message
#> <char>
#> 1: Group has less than 2 individuals after applying 'reference'.
#> 2: Group has less than 2 individuals after applying 'reference'.
#> 3: Group has less than 2 individuals after applying 'reference'.
#> 4: Group has less than 2 individuals after applying 'reference'.
#> 5:
#> 6:9.2 Corrected mean coancestry
(scale = "coancestry")
When scale = "coancestry", pedrel() returns
the diagonal-corrected population mean coancestry following Caballero
& Toro (2000):
where
is the mean off-diagonal relationship,
is the mean inbreeding coefficient of the group, and
is the group size. This
includes both off-diagonal pairwise coancestry and diagonal
self-coancestry. It matches the internal coancestry quantity used by
pediv() to derive
.
coan_by_gen <- pedrel(tp_small, by = "Gen", scale = "coancestry")
coan_by_gen
#> Gen NTotal NUsed MeanCoan Status Message
#> <int> <int> <int> <num> <char> <char>
#> 1: 1 9 9 0.05555556 ok
#> 2: 2 5 5 0.18000000 ok
#> 3: 3 7 7 0.13775510 ok
#> 4: 4 3 3 0.20833333 ok
#> 5: 5 2 2 0.27148438 ok
#> 6: 6 2 2 0.39550781 okUse scale = "relationship" to track mean pairwise
relatedness trends. Use scale = "coancestry" when you need
a population-level coancestry consistent with diversity metrics from
pediv().
pedrel(scale = "coancestry") applies the same
diagonal-corrected formula as pediv(). The resulting
MeanCoan values are therefore directly comparable to
pediv()$summary$MeanCoan, and
GeneDiv = 1 - MeanCoan translates the trend table from
pedrel() into a retained-diversity timeline without any
additional computation.
MeanRel and coancestry-based Ne are
conceptually linked, but they are not identical summaries.
MeanRel is an average additive relationship within a group,
whereas coancestry-based Ne is derived from the rate of
increase in coancestry across pedigree depth.
10. Inbreeding Trends with inbreed() and
pedfclass()
Wright’s inbreeding coefficient (Wright, 1922, 1931) is the probability that the two alleles of an individual are identical by descent. A practical starting point is to inspect the mean trend by generation.
tp_inbred_f <- inbreed(tp_inbred)
f_trend <- tp_inbred_f[, .(MeanF = mean(f, na.rm = TRUE)), by = Gen]
f_trend
#> Gen MeanF
#> <int> <num>
#> 1: 1 0.0000
#> 2: 2 0.0000
#> 3: 3 0.2500
#> 4: 4 0.2500
#> 5: 5 0.4375For classification by inbreeding severity, pedfclass()
can be applied directly to the pedigree object. If the inbreeding
coefficient is not yet present, the function computes it internally.
pedfclass(tp_inbred)
#> Calculating inbreeding coefficients...
#> Key: <FClass>
#> FClass Count Percentage
#> <ord> <int> <num>
#> 1: F = 0 4 57.14286
#> 2: 0 < F <= 0.0625 0 0.00000
#> 3: 0.0625 < F <= 0.125 0 0.00000
#> 4: 0.125 < F <= 0.25 2 28.57143
#> 5: F > 0.25 1 14.28571Custom reporting thresholds can be supplied through
breaks.
pedfclass(tp_inbred, breaks = c(0.03125, 0.0625, 0.125, 0.25))
#> Calculating inbreeding coefficients...
#> Key: <FClass>
#> FClass Count Percentage
#> <ord> <int> <num>
#> 1: F = 0 4 57.14286
#> 2: 0 < F <= 0.03125 0 0.00000
#> 3: 0.03125 < F <= 0.0625 0 0.00000
#> 4: 0.0625 < F <= 0.125 0 0.00000
#> 5: 0.125 < F <= 0.25 2 28.57143
#> 6: F > 0.25 1 14.28571The default thresholds correspond approximately to familiar pedigree scenarios:
- : half-sib mating.
- : avuncular or grandparent-grandchild mating.
- : full-sib or parent-offspring mating.
11. Gene Flow and Partial Inbreeding
11.1 pedancestry(): founder-line proportions
pedancestry() tracks how founder groups contribute to
later descendants. This is useful when founders are labeled by line,
strain, or geographic origin.
ped_line <- data.table(
Ind = c("A", "B", "C", "D", "E", "F", "G"),
Sire = c(NA, NA, NA, NA, "A", "C", "E"),
Dam = c(NA, NA, NA, NA, "B", "D", "F"),
Sex = c("male", "female", "male", "female", "male", "female", "male"),
Line = c("Line1", "Line1", "Line2", "Line2", NA, NA, NA)
)
tp_line <- tidyped(ped_line)
anc <- pedancestry(tp_line, foundervar = "Line")
anc
#> Ind Line1 Line2
#> <char> <num> <num>
#> 1: A 1.0 0.0
#> 2: B 1.0 0.0
#> 3: C 0.0 1.0
#> 4: D 0.0 1.0
#> 5: E 1.0 0.0
#> 6: F 0.0 1.0
#> 7: G 0.5 0.511.2 pedpartial(): which ancestors explain
inbreeding?
pedpartial() decomposes total inbreeding into
contributions from selected ancestors. It is useful for identifying
which ancestors are most responsible for the observed inbreeding.
partial_f <- pedpartial(tp_inbred, ancestors = c("A", "B"))
#> Calculating partial inbreeding for 2 ancestors...
partial_f
#> Ind A B
#> <char> <num> <num>
#> 1: A 0.00000 0.00000
#> 2: B 0.00000 0.00000
#> 3: C 0.00000 0.00000
#> 4: D 0.00000 0.00000
#> 5: E 0.12500 0.12500
#> 6: F 0.00000 0.25000
#> 7: G 0.15625 0.2812512. Practical Interpretation
One useful interpretation order is:
- Check pedigree depth and completeness with
pedecg(). - Check generation timing with
pedgenint(). - Quantify static diversity with
fe,fa,fg, andGeneDivviapediv(); compare withfeHandfaHto assess long-tail founder value. UseGeneDiv(= ) as the headline retained-diversity index for management reporting. - Track diversity dynamics over time with
pedhalflife(). Inspect , , and to identify the dominant driver of loss; use as a headline indicator for management reporting. - Compare demographic, inbreeding, and coancestry-based
Ne. - Monitor
MeanRelandMeanFover time. - Use
pedsubpop(),pedancestry(), andpedpartial()to diagnose structure, introgression, and bottlenecks.
References
- Boichard, D., Maignel, L., & Verrier, É. (1997). The value of using probabilities of gene origin to measure genetic variability in a population. Genetics Selection Evolution, 29(1), 5-23.
- Caballero, A., & Toro, M. A. (2000). Interrelations between effective population size and other pedigree tools for the management of conserved populations. Genetical Research, 75(3), 331-343.
- Cervantes, I., Goyache, F., Molina, A., Valera, M., & Gutiérrez, J. P. (2011). Estimation of effective population size from the rate of coancestry in pedigreed populations. Journal of Animal Breeding and Genetics, 128(1), 56-63.
- Gutiérrez, J. P., Cervantes, I., Molina, A., Valera, M., & Goyache, F. (2008). Individual increase in inbreeding allows estimating effective sizes from pedigrees. Genetics Selection Evolution, 40(4), 359-378.
- Gutiérrez, J. P., Cervantes, I., & Goyache, F. (2009). Improving the estimation of realized effective population sizes in farm animals. Journal of Animal Breeding and Genetics, 126(4), 327-332.
- Hill, M. O. (1973). Diversity and evenness: a unifying notation and its consequences. Ecology, 54(2), 427-432.
- Jost, L. (2006). Entropy and diversity. Oikos, 113(2), 363-375.
- Lacy, R. C. (1989). Analysis of founder representation in pedigrees: founder equivalents and founder genome equivalents. Zoo Biology, 8(2), 111-123.
- Wright, S. (1922). Coefficients of inbreeding and relationship. The American Naturalist, 56(645), 330-338.
- Wright, S. (1931). Evolution in Mendelian populations. Genetics, 16(2), 97-159.